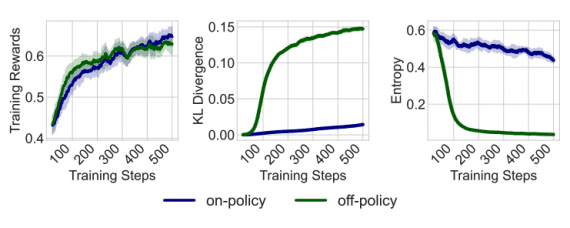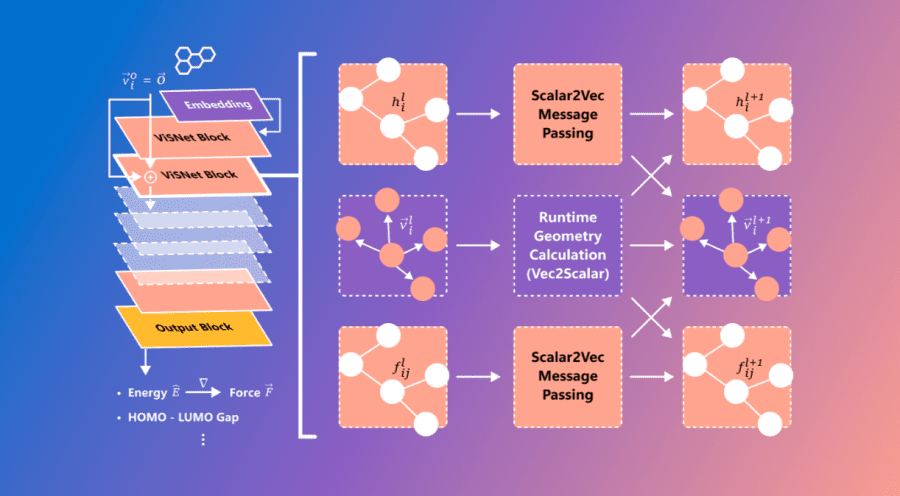
Microsoft has unveiled ViSNet – a graph neural network modeling the geometry of complex molecules to predict their activity. ViSNet has the potential to significantly expedite the search for and study of new drugs.
Modeling molecular geometry enables the prediction of how molecular configurations impact processes such as drug interactions, chemical reactivity, and protein functionality. If feasible, scientists could forecast a drug’s effectiveness as well as its side effects and toxicity before human trials. The key challenge in such modeling lies in the exponentially growing computational costs as molecule size increases.
Traditional molecular dynamics modeling accounts for bond lengths, bond angles, and dihedral angles. The graph neural network VisNet extends these features to interactions, including atom pairs (two-body), atom triplets (three-body), and atom quadruplets (four-body). The algorithm’s complexity is linear, significantly reducing computational complexity.
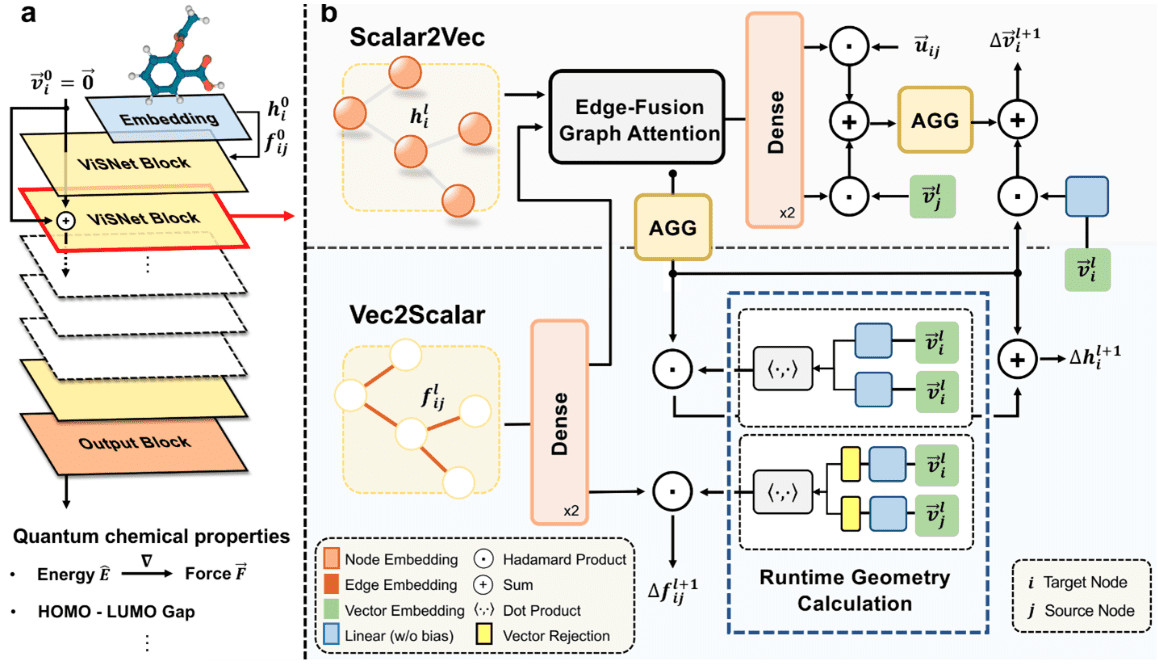
ViSNet was evaluated using standardized criteria for predicting molecular properties. Across all existing datasets (MD17, MD22, QM9, and Molecule3D), ViSNet outperformed existing algorithms in accurately determining molecular geometry. Additionally, ViSNet was utilized in the first global competition for AI-driven drug development, an international competition on predicting inhibitors against the main protease of SARS-CoV-2, incorporating information on small molecule sequences. The competition attracted 1105 participants from 878 teams worldwide, with ViSNet emerging as the victor.
ViSNET has been incorporated into PyTorch as a base model for molecular modeling and property prediction. The model is also published on GitHub.