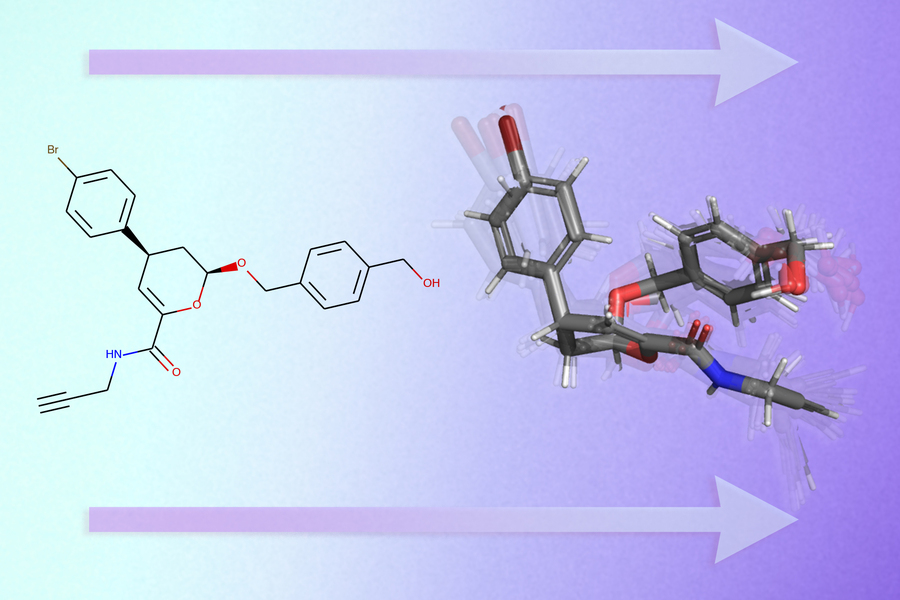
MIT researchers have developed a GeoMol neural network that predicts the three-dimensional structure of a molecule from its two-dimensional graph. This method can accelerate the development of new drugs.
When developing new drugs, scientists are looking for molecules that can attach to disease-causing proteins and change their functionality. To do this, it is important to know the three-dimensional shape of the molecule in order to understand how it will attach to certain protein surfaces. Experimental work in this direction is time-consuming and expensive due to the large number of spatial configurations of molecules.
To solve this problem, MIT has developed a model that predicts the three-dimensional shape of a molecule based on a graph of its molecular structure. Modeling a single molecule using GeoMol takes several seconds and is more accurate than using existing commercial AI tools. GeoMol can help pharmaceutical companies speed up the drug discovery process by reducing the number of molecules they need to test in laboratory experiments.
GeoMol uses the method of transmitting messages by graph. The researchers adapted a neural network to predict specific parameters of molecular geometry. Based on the graph, GeoMol predicts the lengths of chemical bonds between atoms and the angles of these individual bonds. These characteristics determine the possible spatial configurations of the molecule.


