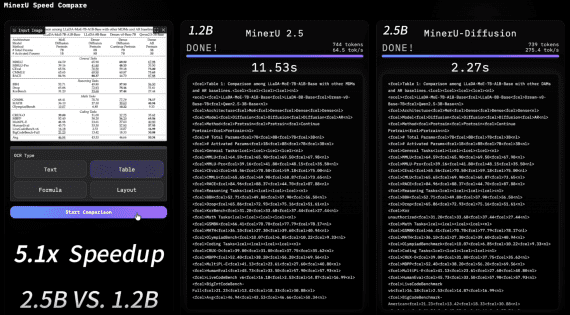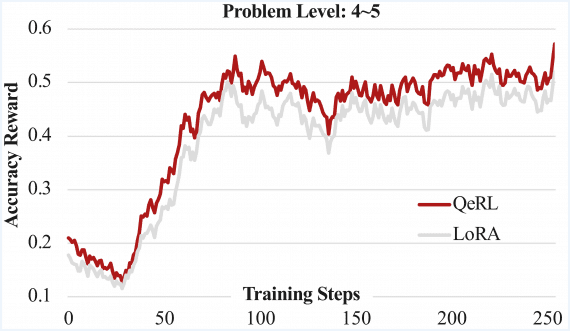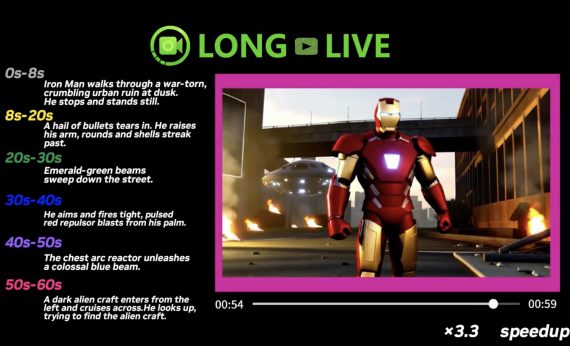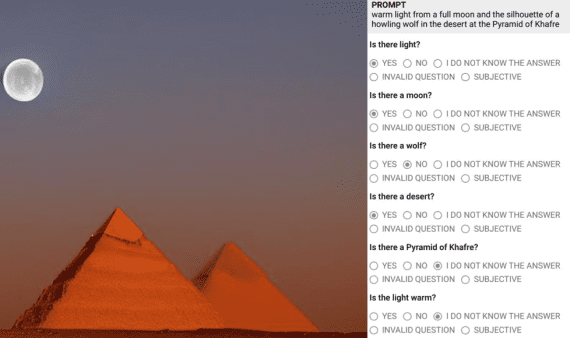
MIT scientists have developed a model that predicts the likelihood of a molecule reaching a transition state—critical for determining the probability of a chemical reaction. Furthermore, researchers will use the model in studies of reactions and catalysts to develop new types of fuels and medications. Therefore, this innovation holds great potential for the chemical industry.
Density functional theory methods traditionally perform the computation of transition states. However, these calculations require vast computational power and take several days for a single state. Contrastingly, the MIT-developed model takes only seconds to compute, marking a significant advancement.
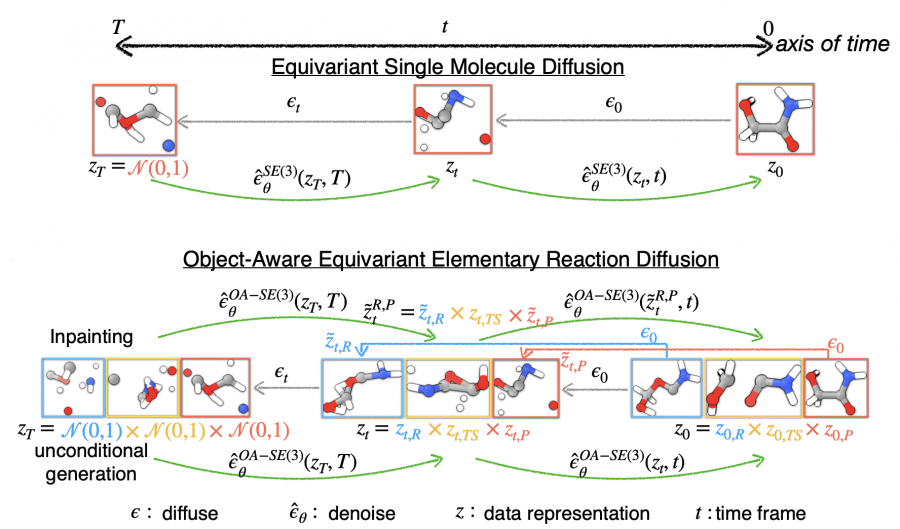
The novelty lies in the fact that the reactive molecules of a hypothetical reaction do not need a predefined orientation relative to each other before entering the model—the model itself determines the orientation that maximizes the probability of existing in the transition state. Consequently, the model adds a layer of efficiency in simulating chemical reactions.
For their diffusion model, scientists utilized the structures of reactants, products, and transition states, calculated using quantum computing methods for 9,000 different chemical reactions. As a result, the model is grounded on a robust dataset, enhancing its predictive power.
![]() Testing the model on 1,000 reactions it had not seen during training, the model’s predictions aligned with the transition state structures generated using quantum methods to an accuracy of 0.08 angstroms. This accuracy highlights the model’s remarkable potential for practical applications.
Testing the model on 1,000 reactions it had not seen during training, the model’s predictions aligned with the transition state structures generated using quantum methods to an accuracy of 0.08 angstroms. This accuracy highlights the model’s remarkable potential for practical applications.
Although scientists primarily trained the model on reactions involving compounds with a relatively small number of atoms—up to 23 atoms for the entire system—they discovered that the model could also provide accurate predictions for reactions involving larger molecules. Thus, the model demonstrates its versatility and scalability.
Details of the training and assessment of the model are outlined in the paper.