A team of researchers from Biohub published ESM Cambrian (ESMC) — a language model for protein structure prediction and design that outperformed AlphaFold3 by Google on structure prediction accuracy, designed molecules that tightly bind to cancer drug targets, and built an atlas of 6.8 billion protein sequences — a ready-made database for discovering new drugs, enzymes, and materials without costly laboratory experiments. On the FoldBench benchmark, the model achieved a DockQ pass rate of 50% on the antibody-antigen complex prediction task from a single sequence, surpassing AlphaFold3’s 47% — while AlphaFold3 relies on MSA (multiple sequence alignment), an alignment of hundreds of evolutionarily related sequences that requires expensive search across billions of records. The project is fully open: weights and code are available on GitHub and HuggingFace, and an interactive environment is running at biohub.ai.
Model specifications:
- ESMC 300M — 300M parameters, 30 transformer layers, embedding dimension 960;
- ESMC 600M — 600M parameters, 36 layers, embedding dimension 1152;
- ESMC 6B — 6B parameters, 80 layers, embedding dimension 2560. Training set — 2.8B protein sequences;
- ESMFold2 — 48 layers, predicts structure from 1024 residues in 15.8 sec on H100;
- ESMFold2-Fast — 24 layers, same task in 9.4 sec on H100.
Architecture: a transformer on 2.8 billion protein sequences
ESMC is a family of transformer models trained on the masked token prediction objective: a portion of amino acids in a sequence is hidden, and the model learns to restore them. The same approach is used in language models for text: models like BERT learned to understand word meaning and relationships simply by filling in gaps — without dictionaries, grammar books, or labeled data. ESMC does the same with proteins.
Training was conducted at three scales: 300 million, 600 million, and 6 billion parameters. The key difference from the previous generation (ESM2) is that the authors added metagenomic sequences collected from soil, ocean, and animal gut samples, expanding the training set 56-fold: from ~50 million to ~2.8 billion sequences. This produced a clear scaling law: a log-linear improvement in representation quality with FLOPs growth (R² = 0.99), with no plateau even at the transition from 650 million to 6 billion parameters — unlike ESM2, where returns from scaling diminished.
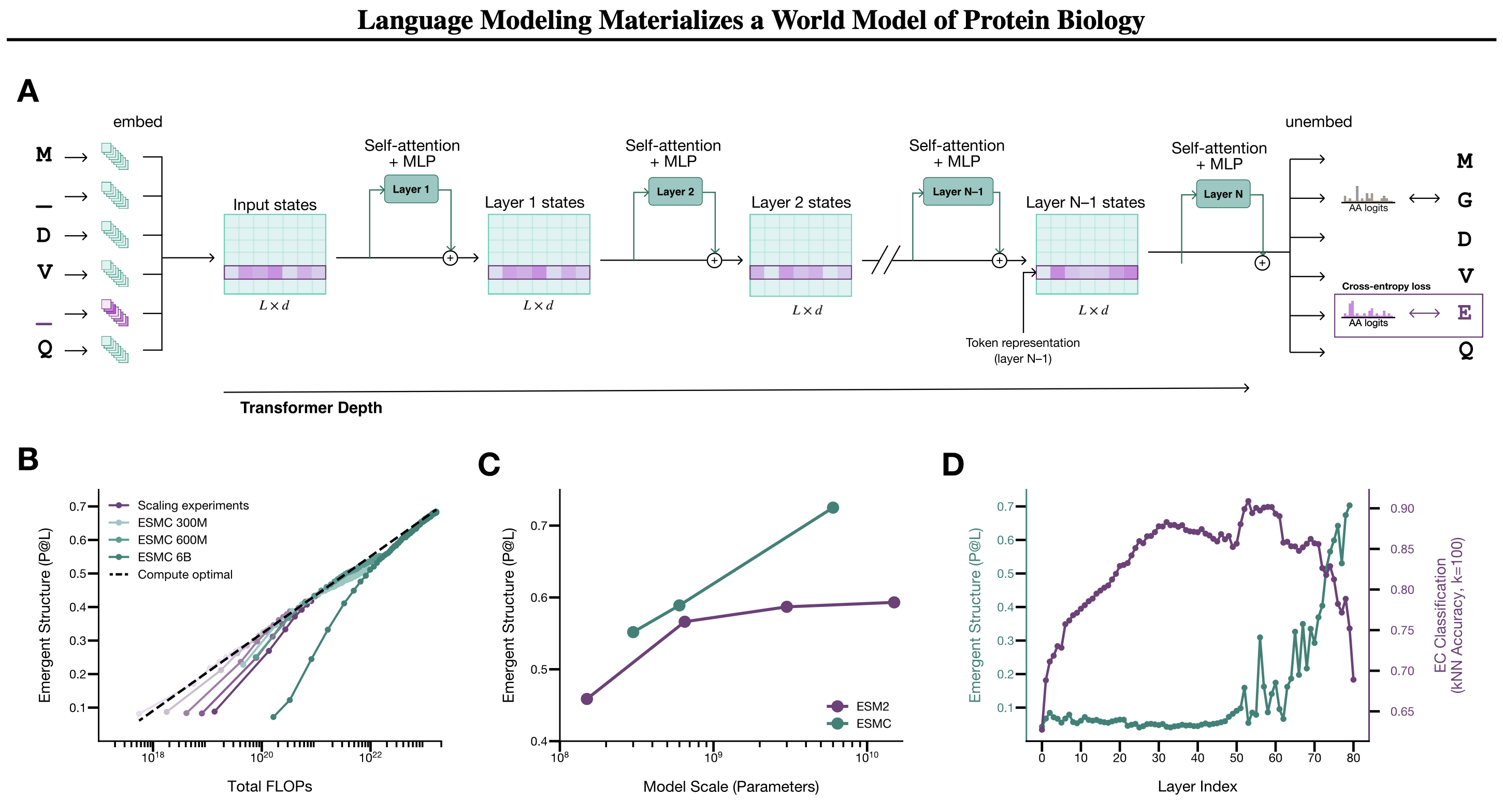
Each sequence position is encoded as a vector of dimension 2560 (for the 6B model). This is the standard transformer idea: each token becomes a point in a high-dimensional space, where nearby points indicate similar context.
Structural and functional information about a protein is distributed across different layers of the network. If you train a linear classifier on the activations of each layer and measure how well it predicts protein function and 3D structure, the picture is clear: functional features are best read at layers 50–60 (around 3/4 of network depth), structural features — at the very last layers. The model did not simply memorize sequences; it organized its internal representations hierarchically: abstract function first, concrete geometry later. This is why the authors use layer 60 representations for interpretability tasks and final layers for structure prediction.
ESMFold2: how to build the world’s best structure predictor from a frozen encoder
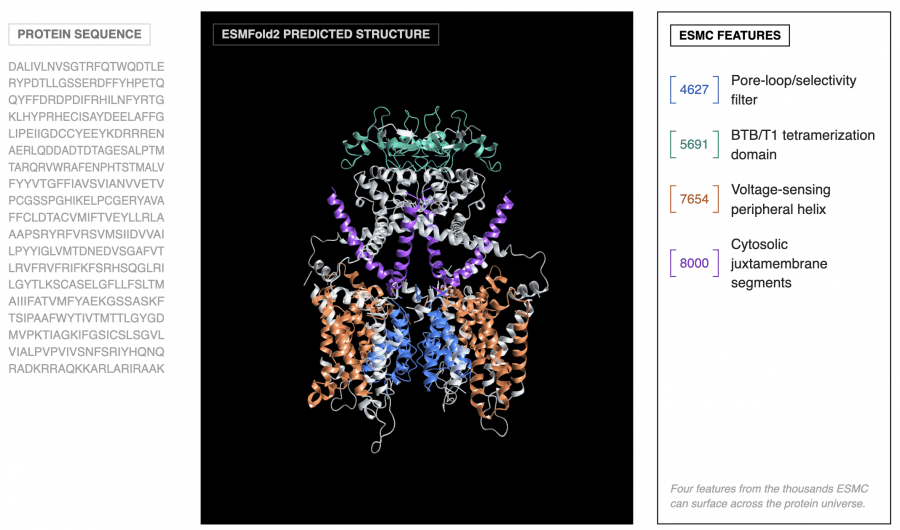
Building on ESMC, the authors developed ESMFold2 — a model for predicting the 3D structure of proteins and their complexes. The architectural idea is elegant: the weights of ESMC 6B are fully frozen, and a separate folding head is trained on top. Representations from all 80 ESMC layers are merged into a two-dimensional pairwise representation — a matrix of interactions between all pairs of positions. This representation is then iteratively refined through 48 recurrent folding layers with a stabilized recurrent update, enabling backpropagation through the recursive loop. The final stage is a diffusion transformer with sliding-window atom attention that denoises Gaussian noise into atomic coordinates.
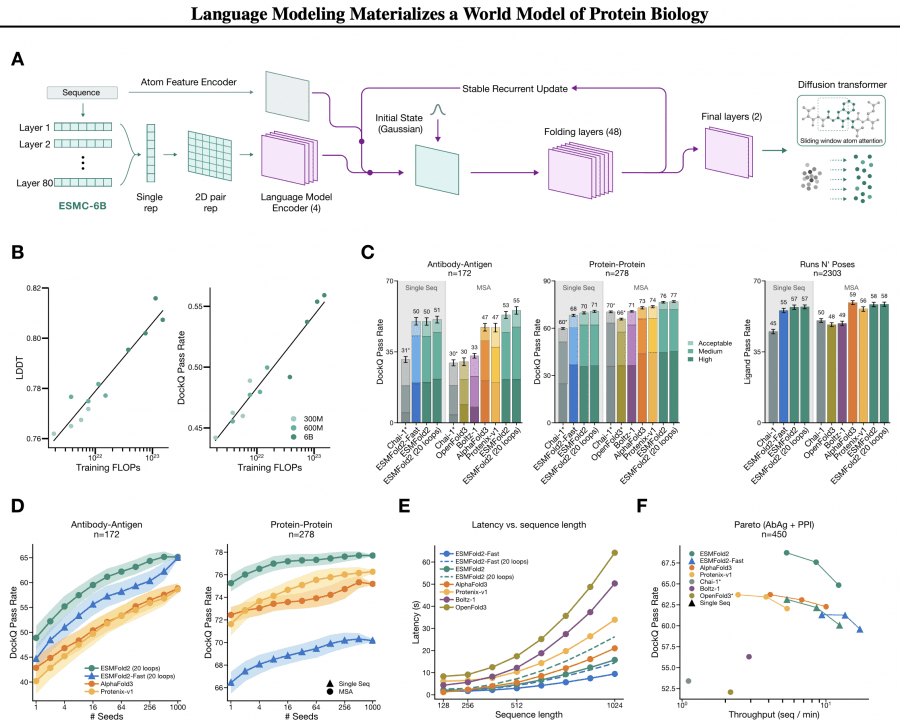
The FoldBench results speak for themselves. ESMFold2 predicts antibody-antigen complexes with a DockQ pass rate of 50% ± 2% from a single sequence without MSA, while AlphaFold3 with MSA achieves 47% ± 2%. With MSA, ESMFold2 reaches 53% (antibody-antigen) and 76% (protein-protein). On speed: a structure from 1024 residues is predicted in 15.8 seconds on a single H100 — 1.3× faster than AlphaFold3. The lightweight ESMFold2-Fast with 24 layers instead of 48 handles the same task in 9.4 seconds while still outperforming AlphaFold3 on accuracy.
On inference-time scaling: with 1000 samples ranked by iPTM score, accuracy on the antibody-antigen task grows from 49% to 65%. ESMFold2-Fast at 1000 samples matches the full model (65% vs 65%) — meaning a smaller model with a larger inference compute budget achieves the same result as a larger model in a single run. This directly follows from the diffusion transformer’s sample diversity genuinely covering the solution space.
Differentiable design: backpropagation through the model to find therapeutic molecules
The most practically significant application is de novo protein binder design. The idea is to search for the optimal sequence directly in the input space of the combined ESMC + ESMFold2 model. Formally, this is a joint optimization of a candidate sequence x and its structure s given a fixed target sequence t:
p(x, s) = p(s | x, t) · p(x)
where p(x) is a sequence prior from ESMC and p(s | x, t) is the conditional structure model ESMFold2. Technically, sequences are represented as continuous distributions over amino acid probabilities, optimization runs via backpropagation through ESMC and ESMFold2, and temperature annealing gradually sharpens the distribution into discrete sequences. No RL, no diffusion-based generation — a single-step differentiable optimization.
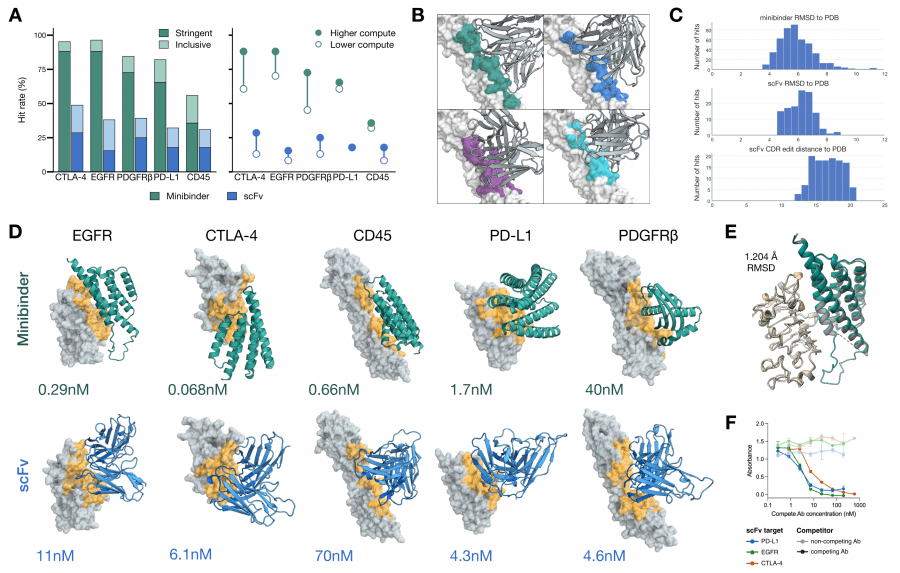
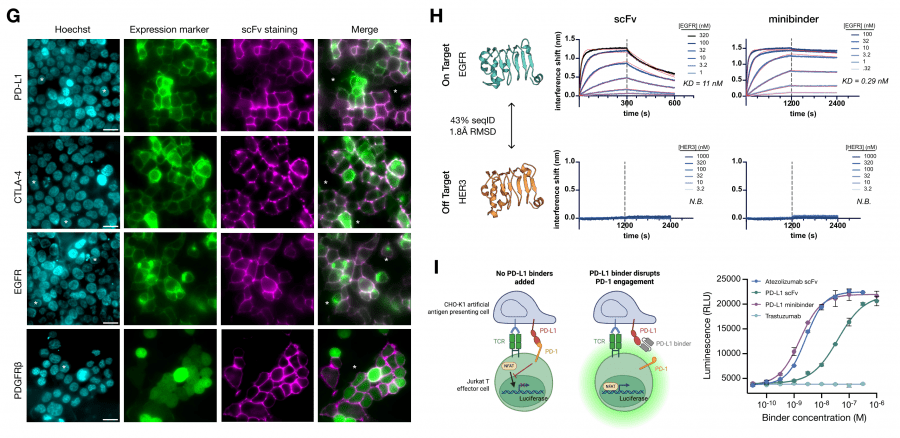
Design was conducted for five oncology targets: EGFR, CTLA-4, CD45, PD-L1, and PDGFRβ. For each target, two types of molecules were created: compact minibinders (de novo, without an immunoglobulin scaffold) and scFv (single-chain variable fragments retaining the immunoglobulin scaffold with optimized CDR loops).
Experimental validation by BLI showed: minibinder for CTLA-4 — KD = 0.068 nM, for EGFR — 0.29 nM, for CD45 — 0.66 nM. Among scFv: CTLA-4 — 6.1 nM, PD-L1 — 4.3 nM, EGFR — 11 nM. For PD-L1, the authors confirmed that the designed minibinder blocks the PD-L1/PD-1 interaction and activates T cells in a functional assay at a level comparable to clinically approved antibodies. The predicted minibinder structure matched the crystal structure with RMSD 1.204 Å — meaning the model predicted not just a binding sequence but also the correct 3D conformation.
What the transformer learned: sparse autoencoders reveal the model’s internal concepts
This is arguably the most interesting section of the paper from an ML perspective. The authors applied the mechanistic interpretability technique from the LLM world to a protein model and got a surprisingly clear picture.
The problem is standard: neurons in language models are polysemantic — a single neuron activates for many unrelated concepts simultaneously (superposition). To extract monosemantic features, sparse autoencoders (SAEs) are trained: they project each amino acid vector into a sparse high-dimensional space where each dimension corresponds to one concept. SAEs were trained on 8 billion tokens for each layer of ESMC 300M, 600M, and 6B, with feature spaces from 2¹³ to 2¹⁷ and varying sparsity levels.
Feature hierarchy in ESMC’s latent space:
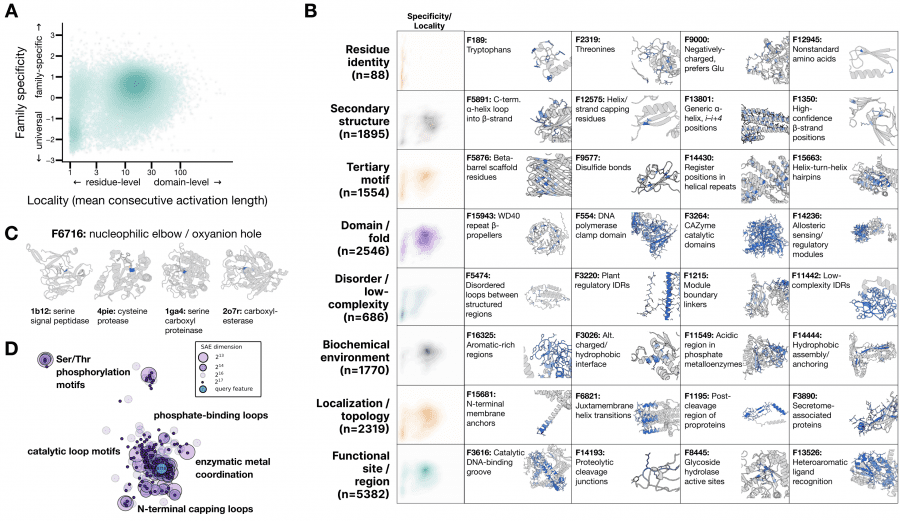
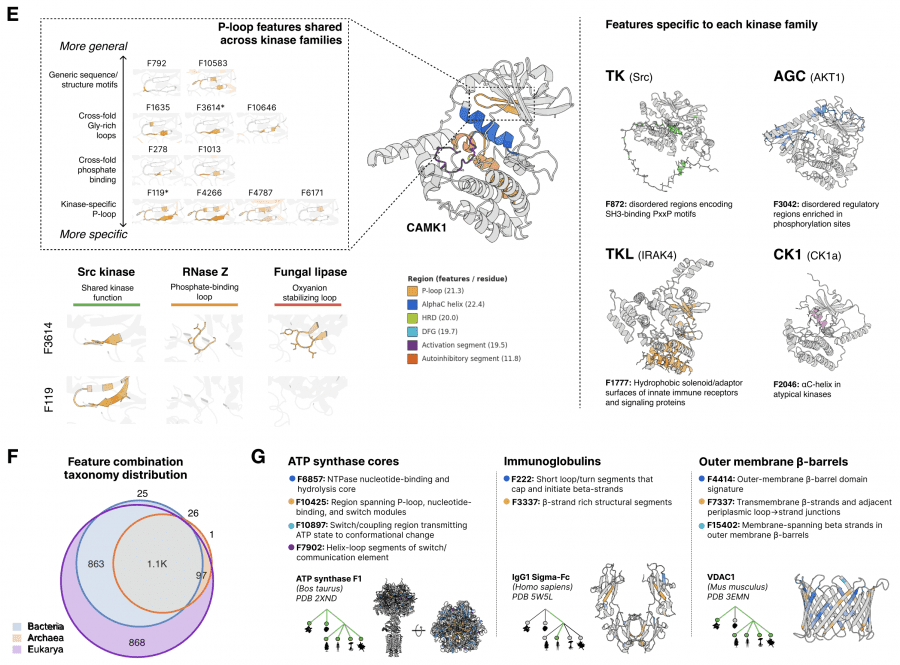
For feature interpretation, an agentic pipeline was used: an AI agent analyzed 195,000 annotated proteins from SwissProt and produced a natural-language description for each of the 16,384 features. The result is striking: the features form a hierarchy that precisely matches how biologists describe proteins — from individual amino acid identity to evolutionary themes shared across entire domains of life. No biological labels were used during ESMC training. This is a clean example of emergence: the pattern arose on its own during training without any supervision.
A telling example: one SAE feature (F6716) activates on the nucleophilic elbow — a catalytic motif that positions the reactive atom in enzymes. This motif evolved independently in dozens of protein families with completely different 3D structures. Yet F6716 fires on it in 75 of 99 relevant enzymes, spanning 25 distinct structural topologies. The model independently discovered this biochemical principle.
A key practical property: a linear model built on just 108 SAE features predicts the Src kinase activity landscape around the wild type with Spearman ρ = 0.74. This means SAE features are not just descriptive — they predict how point mutations will change protein function.
Atlas of the protein world: UMAP over 7.7 million clusters
Using SAE embeddings as a universal vector representation, the authors built an atlas of all protein diversity on Earth. Data was collected from eight public databases: UniParc, SPIRE, MGnify, UHGG, and four JGI datasets — totaling 6.8 billion unique sequences, of which 5.6 billion came from metagenomic samples of soil, ocean, and gut.
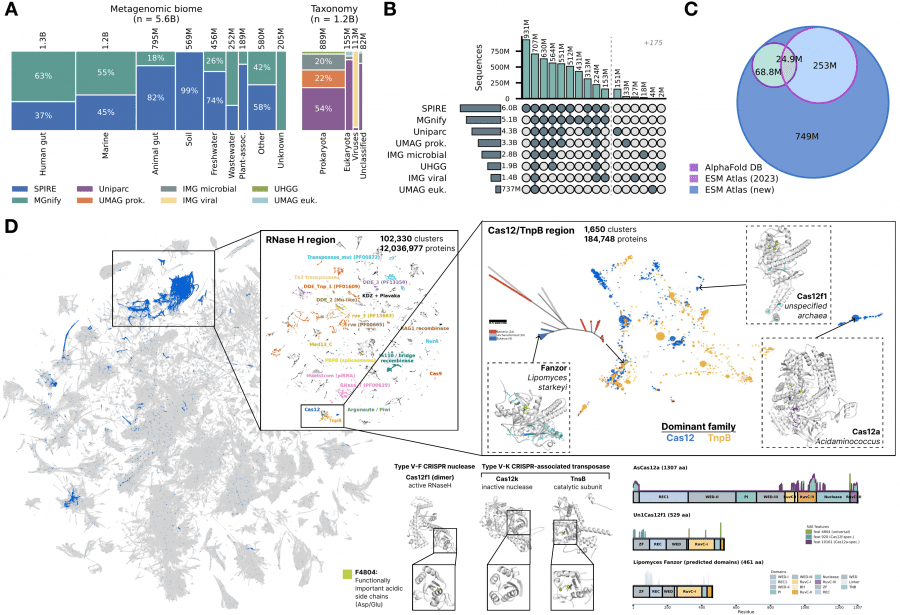
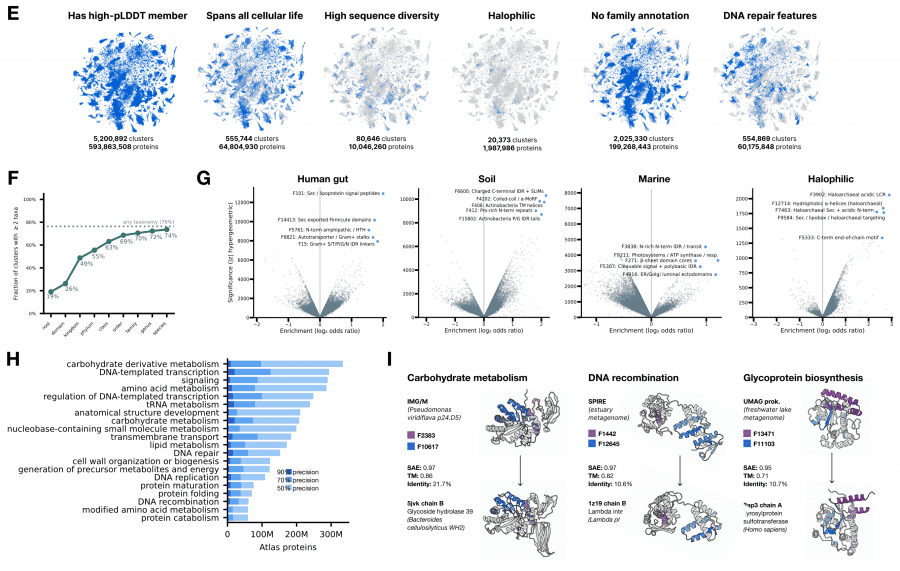
ESMFold2 predicted structures for 1.1 billion representative sequences — 835 million more than the AlphaFold Database, and 756 million of them have no structure prediction in any existing database. Clustering by SAE similarity (Jaccard threshold 0.6) yielded 230 million clusters, of which 7.7 million contain more than 50 members.
The key technical result: SAE similarity outperforms sequence and structure search for finding proteins with shared enzymatic function, especially when sequence identity falls below 40%. This means the SAE feature space encodes functional similarity better than direct sequence or structure comparison — the model learned to represent what a protein does, not just what it looks like.
On the UMAP projection of 7.7 million clusters, the Cas12 family and its evolutionary ancestor TnpB cluster together, while specific SAE feature activations separate catalytically active and inactive variants within the family. This is practically useful: the atlas can be used as a search engine over protein functional space without needing to know sequences or structures in advance.
Why this matters for understanding language models in general
The paper asks an uncomfortable question: what does a language model actually learn when trained to predict the next token? In the case of proteins, the answer can be verified experimentally. A model trained solely on masked prediction without any biological labels independently reproduced the hierarchical organization of protein biology — the same organization that scientists built over decades of experiments. SAE features precisely correspond to concepts from molecular biology textbooks, and their relative positions in the feature space reflect biological hierarchies.
This is consistent with observations in NLP: large language models for text also form internal representations of grammar and semantics without explicit supervision. But in biology, these representations can be validated experimentally. That is exactly what the authors do — showing that SAE features predict mutational effects, that predicted structures match crystal structures, and that designed molecules actually bind their targets. World model is not a metaphor. It is a verifiable claim.