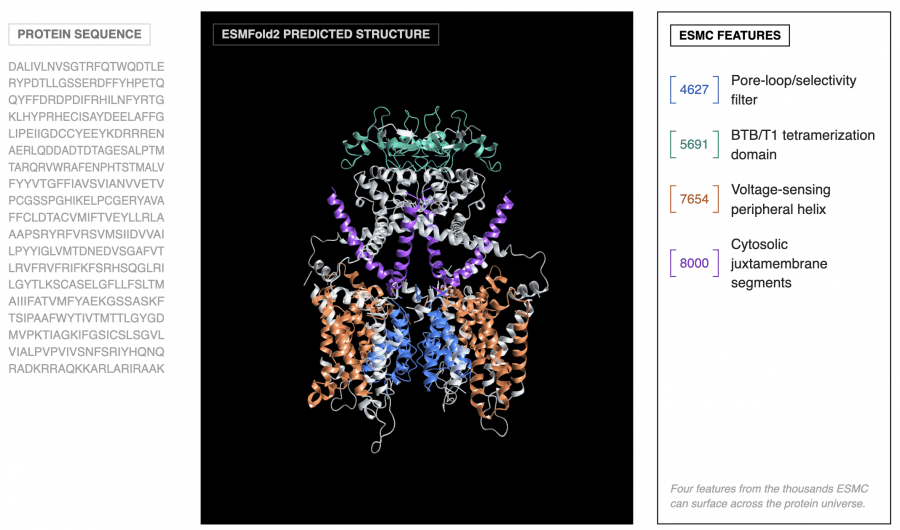
Команда исследователей из Biohub опубликовала ESM Cambrian (ESMC) — языковую модель для предсказания и дизайна белков, которая обошла AlphaFold3 от Google по точности предсказания структур, спроектировала молекулы, которые крепко связываются с белками-мишенями при лечении рака и построила атлас из 6,8 миллиарда белковых последовательностей — готовую базу для поиска новых лекарств, ферментов и материалов без дорогостоящих лабораторных экспериментов. На бенчмарке FoldBench модель достигла DockQ pass rate 50% на задаче предсказания комплекса антитело-антиген из одной последовательности, превзойдя AlphaFold3 с показателем 47%, при том что AlphaFold3 для этого использует MSA (multiple sequence alignment) — выравнивание сотен эволюционно родственных последовательностей, которое требует дорогостоящего поиска по миллиардам записей. Проект полностью открытый: веса и код доступны на GitHub и HuggingFace, интерактивная среда запущена на biohub.ai.
Характеристики модели:
- ESMC 300M — 300 млн параметров, 30 трансформерных слоёв, размерность векторов 960;
- ESMC 600M — 600 млн параметров, 36 слоёв, размерность 1152;
- ESMC 6B — 6 млрд параметров, 80 слоёв, размерность 2560. Обучающая выборка — 2,8 млрд белковых последовательностей;
- ESMFold2 — 48 слоёв, предсказывает структуру из 1024 остатков за 15,8 сек на H100;
- ESMFold2-Fast — 24 слоя, та же задача за 9,4 сек на H100.
Архитектура: трансформер на 2,8 миллиарда белковых последовательностей
ESMC — семейство трансформер-моделей, обученных по принципу masked language modeling: часть аминокислот в последовательности маскируется, и модель учится восстанавливать пропущенные токены. Этот же подход работает для текстов: модель восстанавливает пропущенные слова и в процессе выучивает семантику и грамматику без каких-либо меток. С белками то же самое: восстанавливая пропущенные аминокислоты, модель вынуждена самой разобраться в структуре, функции и эволюционных ограничениях белков.
Обучение проводилось на трёх масштабах: 300 миллионов, 600 миллионов и 6 миллиардов параметров. Главное отличие от предыдущего поколения (ESM2) — авторы добавили метагеномные последовательности, собранные из образцов почвы, океана и кишечника животных, расширив обучающую выборку в 56 раз: с ~50 миллионов до ~2,8 миллиарда последовательностей. Это дало чёткий закон масшабирования: log-линейный прирост качества представлений с ростом FLOPs (R² = 0,99), причём плато не наблюдается даже при переходе с 650 миллионов на 6 миллиардов параметров в отличие от ESM2, где отдача от масштабирования падала.
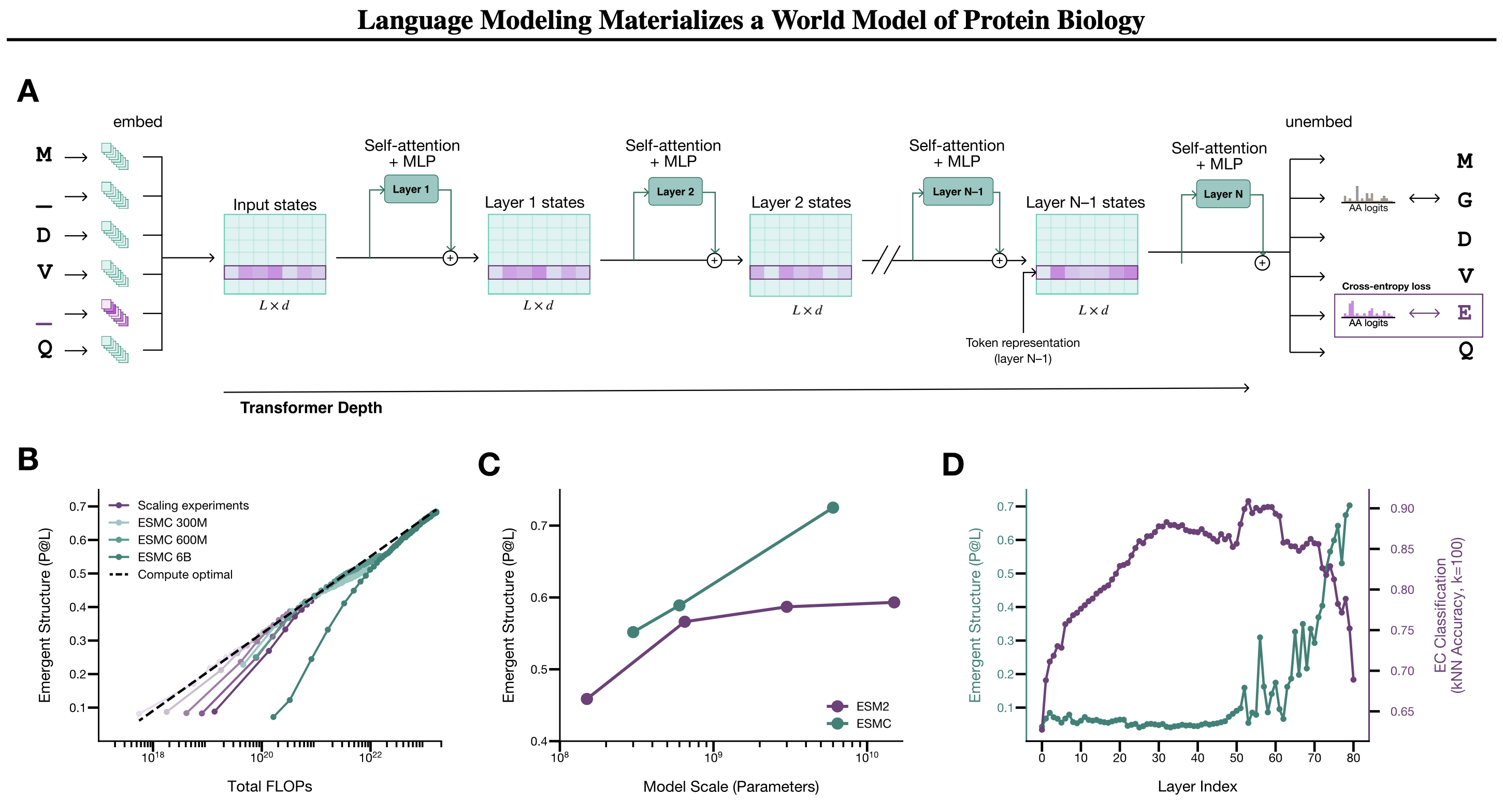
Каждая позиция последовательности кодируется вектором размерностью 2560 (для модели 6B). Это стандартная идея для трансформеров: каждый токен превращается в точку в многомерном пространстве, где близкие точки означают похожий контекст.
Информация о структуре и функции белка распределена по разным слоям сети. Если обучить линейный классификатор на активациях каждого слоя и проверить, насколько хорошо он предсказывает функцию белка и его трёхмерную структуру, картина будет такой: функциональные признаки лучше всего читаются на слоях 50–60 (около 3/4 глубины сети), структурные — в самых последних слоях. Модель не просто запомнила последовательности, а организовала внутренние представления иерархически: сначала абстрактная функция, потом конкретная геометрия. Именно поэтому авторы берут представления со слоя 60 для задач интерпретируемости, а финальные слои используют для предсказания структуры.
ESMFold2: как из замороженного энкодера сделать лучший в мире предсказатель структур
На базе ESMC авторы разработали ESMFold2 — модель для предсказания трёхмерной структуры белков и их комплексов. Архитектурная идея интересная: веса ESMC 6B полностью заморожены, а поверх них обучается отдельная folding-голова. Представления всех 80 слоёв ESMC объединяются в двумерное попарное (pairwise) представление — матрицу взаимодействий между всеми парами позиций. Это представление затем итеративно уточняется через 48 рекуррентных folding-слоёв со стабилизированным обновлением состояния, что позволяет делать обратное распространение через рекурсивный цикл. Финальный этап — диффузионный трансформер со скользящим окном атомарного внимания (sliding-window atom attention), который денойзит гауссовский шум до атомарных координат.
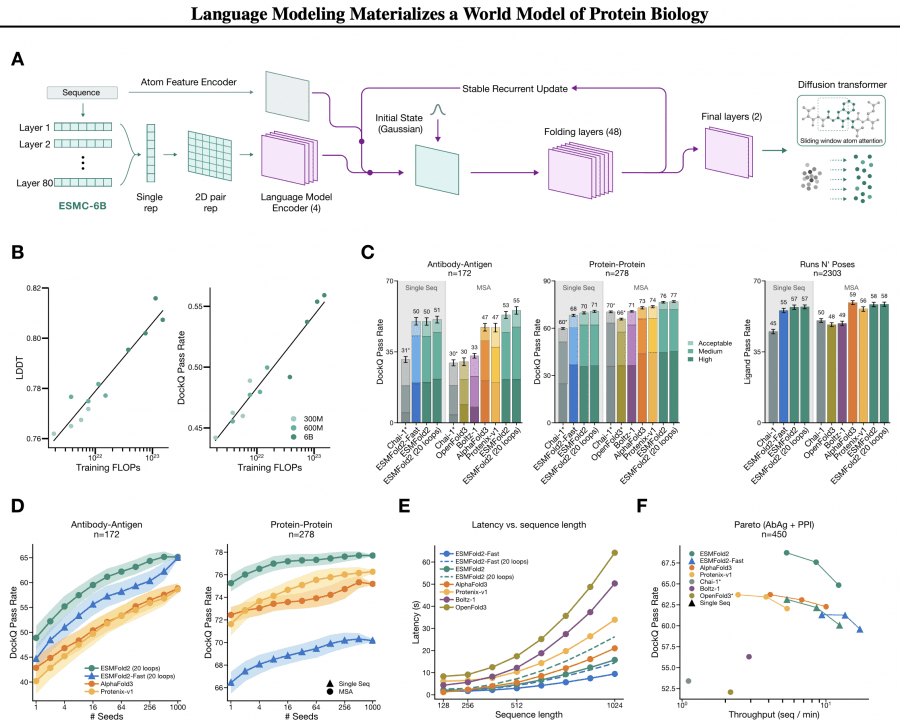
Результаты на FoldBench говорят сами за себя. ESMFold2 предсказывает комплекс антитело-антиген с DockQ pass rate 50% ± 2% из одной последовательности без MSA, тогда как AlphaFold3 с MSA даёт 47% ± 2%. При добавлении MSA ESMFold2 вырастает до 53% (антитело-антиген) и 76% (белок-белок). По скорости: структура из 1024 остатков предсказывается за 15,8 секунды на одном H100 — в 1,3 раза быстрее AlphaFold3. Облегчённая ESMFold2-Fast с 24 folding-слоями вместо 48 справляется с той же задачей за 9,4 секунды, при этом также превосходя AlphaFold3 по точности.
Отдельно про inference-time scaling: при 1000 семплах с ранжированием по iPTM score точность на антитело-антиген задаче вырастает с 49% до 65%. ESMFold2-Fast при 1000 семплах догоняет полную модель (65% vs 65%) — то есть меньшая модель при бо́льшем вычислительном бюджете на inference достигает того же результата, что бо́льшая при одном запуске. Это прямое следствие того, что разнообразие семплов из диффузионного трансформера реально покрывает пространство решений.
Дифференцируемый дизайн: backpropagation сквозь модель для поиска терапевтических молекул
Самое практически важное применение — дизайн белков-связывателей de novo. Идея в том, чтобы искать оптимальную последовательность прямо в пространстве входных данных объединённой модели ESMC + ESMFold2. Формально это совместная оптимизация последовательности-кандидата x и его структуры s при фиксированной последовательности мишени t:
p(x, s) = p(s | x, t) · p(x)
где p(x) — prior по последовательностям от ESMC, а p(s | x, t) — условная структурная модель ESMFold2. Технически последовательности представляются как непрерывные распределения над вероятностями аминокислот, оптимизация идёт через обратное распространение сквозь ESMC и ESMFold2, а temperature annealing постепенно заостряет распределение до дискретных последовательностей. Никакого RL, никаких диффузионных методов генерации — одноэтапная дифференцируемая оптимизация.
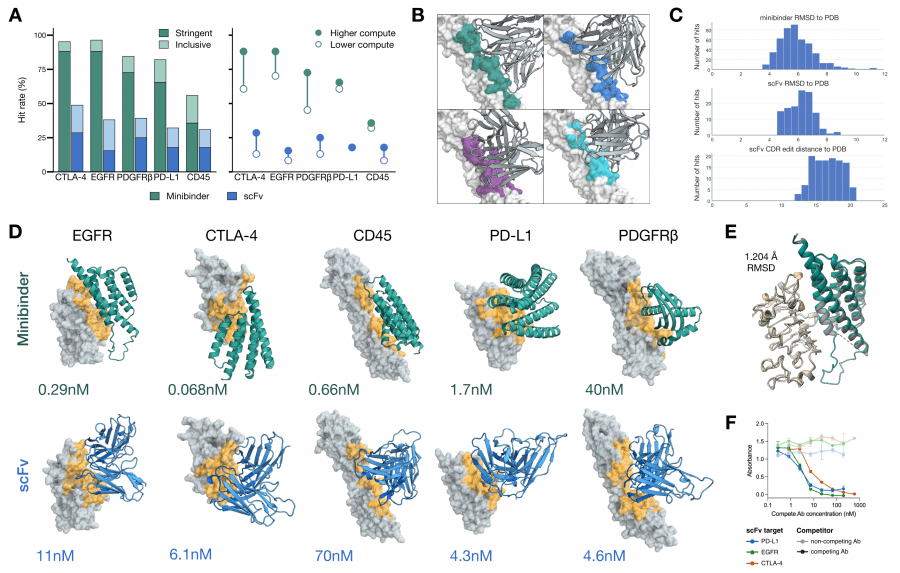
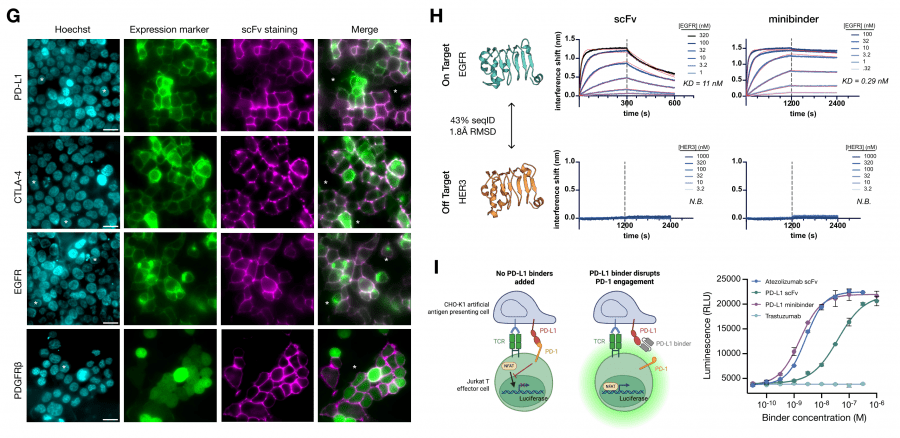
Дизайн проводился для пяти онкологических мишеней: EGFR, CTLA-4, CD45, PD-L1 и PDGFRβ. Для каждой создавались два типа молекул: компактные минибелки (de novo, без иммуноглобулинового каркаса) и scFv (одноцепочечные вариабельные фрагменты антител, сохраняющие иммуноглобулиновый каркас с оптимизированными петлями CDR).
Экспериментальная валидация методом BLI показала: минибелок для CTLA-4 — KD = 0,068 нМ, для EGFR — 0,29 нМ, для CD45 — 0,66 нМ. Среди scFv: CTLA-4 — 6,1 нМ, PD-L1 — 4,3 нМ, EGFR — 11 нМ. Для PD-L1 авторы подтвердили, что спроектированный минибелок блокирует взаимодействие PD-L1/PD-1 и активирует T-клетки в функциональном анализе на уровне, сопоставимом с клинически одобренными антителами. Предсказанная структура минибелка совпала с кристаллической с RMSD 1,204 Å — это значит, что модель предсказала не просто связывающую последовательность, но и правильную трёхмерную конформацию.
Что «понял» трансформер: разреженные автоэнкодеры раскрывают внутренние концепции модели
Это, пожалуй, самый интересный с точки зрения ML раздел статьи. Авторы применили технику mechanistic interpretability из мира LLM к белковой модели и получили неожиданно чёткую картину.
Проблема стандартная: нейроны языковых моделей полисемантичны, то есть один нейрон активируется на множество несвязанных концепций одновременно (superposition). Чтобы извлечь моносемантические признаки, обучаются разреженные автоэнкодеры (SAE): они проецируют вектор каждой аминокислоты в разреженное высокомерное пространство, где каждое измерение отвечает за одну концепцию. SAE обучался на 8 миллиардах токенов для каждого слоя ESMC 300M, 600M и 6B, с пространствами признаков от 2¹³ до 2¹⁷ и разными уровнями разреженности.
Иерархия признаков в латентном пространстве ESMC:
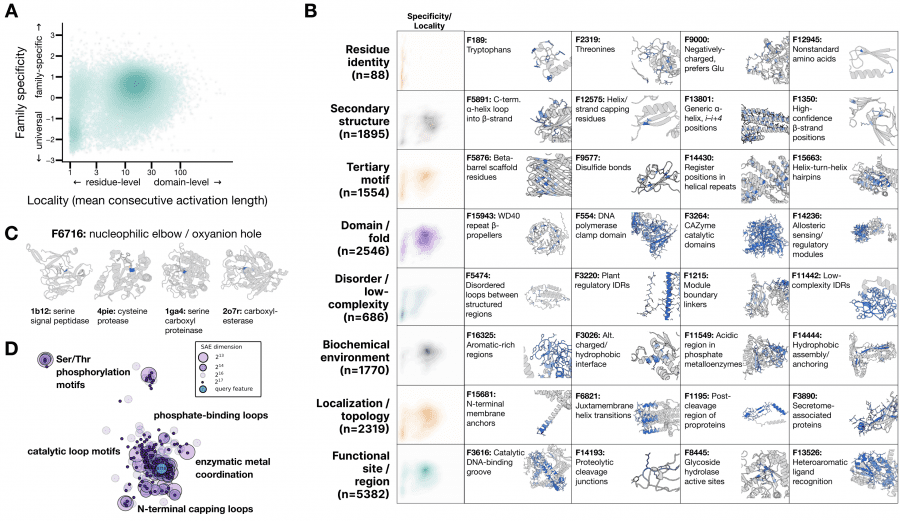
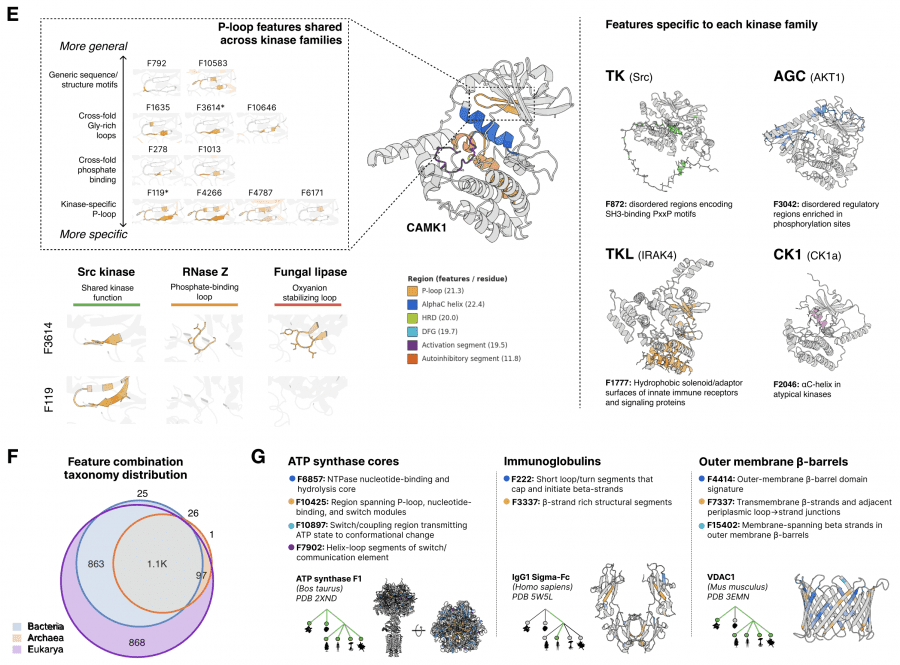
Для интерпретации признаков использовалась агентная система: ИИ-агент анализировал 195 тысяч аннотированных белков из SwissProt и давал текстовое описание каждому из 16 384 признаков. Итог оказался впечатляющим: признаки образуют иерархию, точно соответствующую тому, как биологи описывают белки: от идентичности отдельных аминокислот до эволюционных тем, общих для целых доменов жизни. При этом никакой биологической разметки при обучении ESMC не было, это чистый пример эмерджентности: паттерн возник сам по себе в процессе обучения без какой-либо разметки.
Показательный пример: один признак SAE (F6716) активируется на нуклеофильном локте — каталитическом мотиве, позиционирующем реакционный атом в ферментах. Этот мотив эволюционировал независимо в десятках белковых семейств с совершенно разной трёхмерной структурой. Тем не менее F6716 срабатывает на нём в 75 из 99 релевантных ферментов, охватывая 25 различных структурных топологий. Модель самостоятельно «открыла» этот биохимический принцип.
Важное свойство для практики: линейная модель на основе всего 108 SAE-признаков предсказывает ландшафт активности киназы Src вокруг дикого типа с точностью Spearman ρ = 0,74. Это означает, что SAE-признаки не просто описательны — они предсказывают, как точечные мутации изменят функцию белка.
Атлас белкового мира: UMAP для 7,7 миллиона кластеров
Используя SAE-эмбеддинги как универсальное векторное представление, авторы построили атлас всего белкового разнообразия жизни на Земле. Данные собраны из восьми публичных баз: UniParc, SPIRE, MGnify, UHGG и четыре набора из JGI — суммарно 6,8 миллиарда уникальных последовательностей, из которых 5,6 миллиарда получены из метагеномных образцов почвы, океана и кишечника.
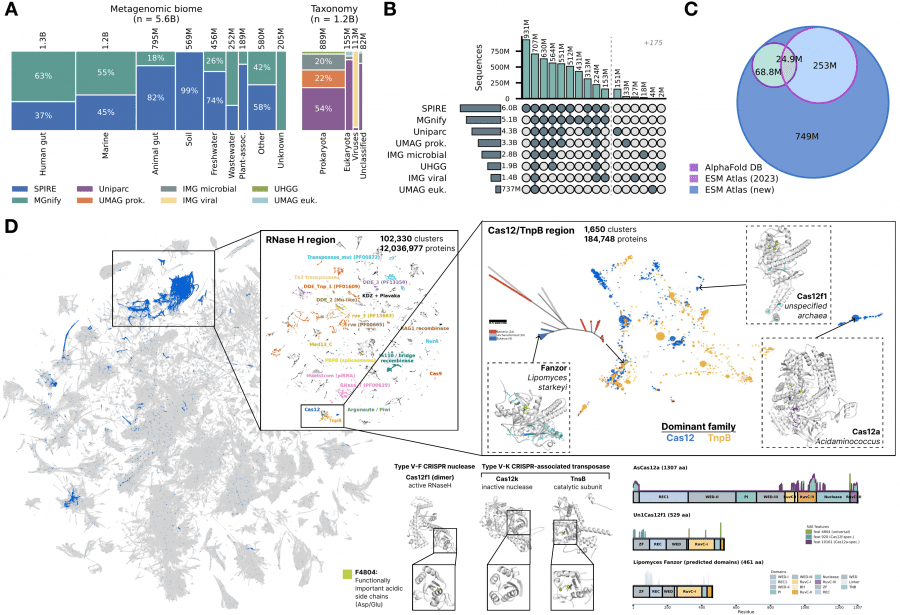
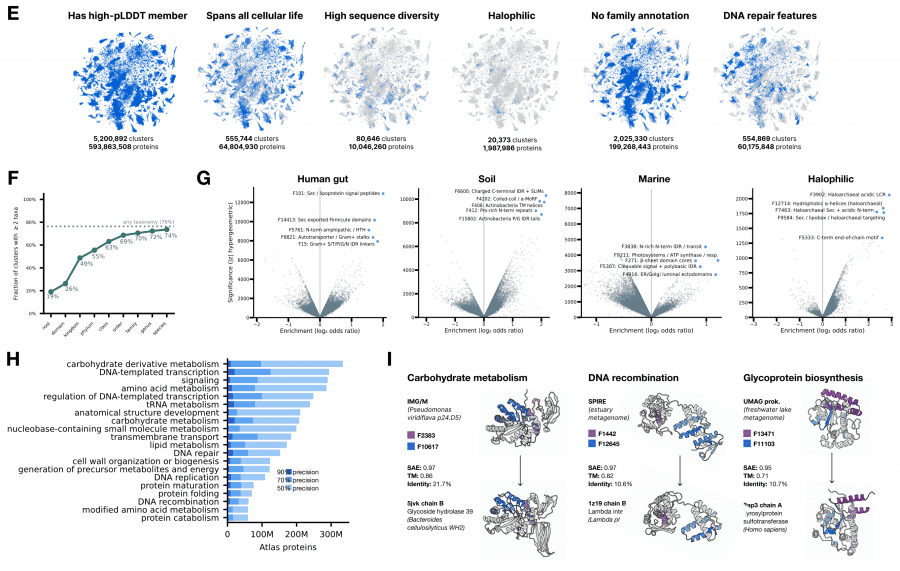
ESMFold2 предсказал структуры для 1,1 миллиарда представительных последовательностей — это на 835 миллионов больше, чем в AlphaFold Database, и 756 миллионов из них вообще не имеют структурного предсказания ни в одной существующей базе данных. Кластеризация по SAE-сходству (порог Jaccard 0,6) дала 230 миллионов кластеров, из которых 7,7 миллиона содержат более 50 членов.
Ключевой технический результат: SAE-сходство превосходит поиск по последовательности и структуре для нахождения белков с общей ферментативной функцией, особенно когда идентичность последовательностей ниже 40%. Это означает, что пространство SAE-признаков кодирует функциональное сходство лучше, чем прямое сравнение последовательностей или структур — то есть модель научилась представлять «зачем» белок существует, а не только «как» он выглядит.
На UMAP-проекции 7,7 миллиона кластеров видно, как семейства Cas12 и его эволюционного предка TnpB располагаются рядом, а активация конкретных SAE-признаков разделяет каталитически активные и неактивные варианты внутри семейства. Это практически применимо: атлас можно использовать как поисковик по функциональному пространству белков без необходимости знать их последовательности или структуры заранее.
Почему это важно для понимания языковых моделей в целом
Статья задаёт неудобный вопрос: что языковая модель «на самом деле» выучивает, когда её обучают предсказывать следующий токен? В случае белков ответ удалось проверить экспериментально. Модель, обученная только на задаче masked prediction без каких-либо биологических меток, самостоятельно воспроизвела иерархическую организацию белковой биологии — ту самую, которую учёные строили десятилетиями экспериментов. SAE-признаки точно соответствуют концепциям из учебников молекулярной биологии, а их взаимное расположение в пространстве отражает биологические иерархии.
Это согласуется с наблюдениями в NLP: большие языковые модели для текстов также формируют внутренние представления грамматики и семантики без явного обучения. Но в биологии эти представления можно валидировать экспериментально. Именно это делают авторы, показывая, что SAE-признаки предсказывают эффект мутаций, что предсказанные структуры совпадают с кристаллическими, что спроектированные молекулы реально связываются с мишенями. World model — не метафора, а верифицируемое утверждение.