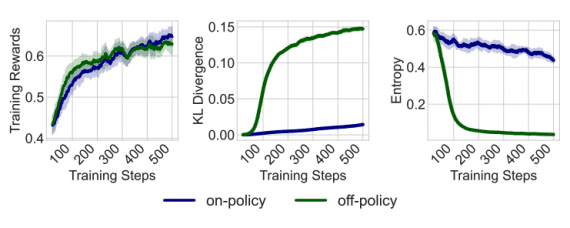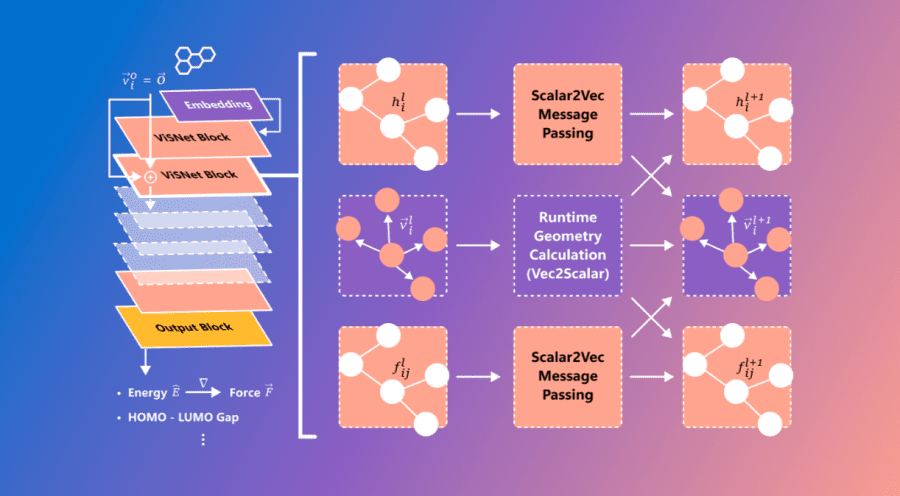
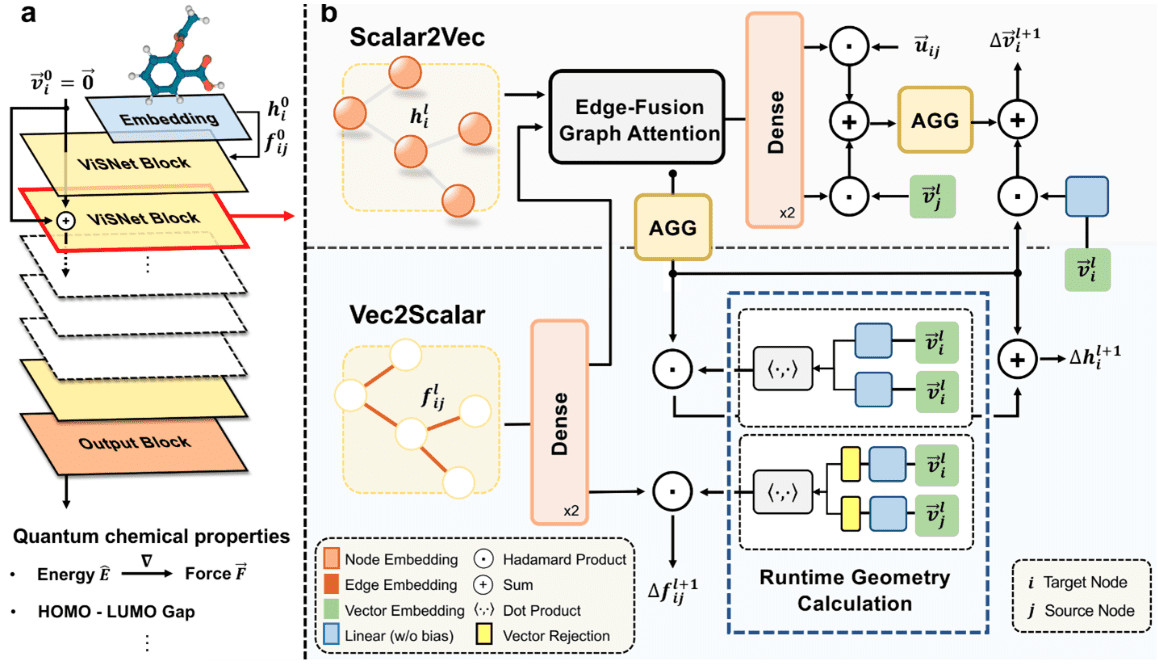
Microsoft опубликовала ViSNet – графовую нейросеть, моделирующую геометрию сложных молекул для предсказания их активности. ViSNet может значительно ускорить поиск и изучение новых лекарств.
Моделирование молекулярной геометрии позволяет предсказывать, как молекулярные конфигурации влияют на такие процессы, как взаимодействие лекарств, химическая реактивность и функциональность белков. Если бы это было возможно, ученые могли бы предсказать эффективность лекарства, а также его побочные эффекты и токсичность до того, как оно будет испытано на людях. Ключевая сложность такого моделирования – экспоненциально растущие вычислительные затраты по мере увеличения размера молекул.
Традиционное моделирование молекулярной динамики учитывает длины связей, углы соединения и двугранные углы. Графовая нейросеть VisNet расширяет эти характеристики на взаимодействия, включающие пары атомов (двухчастичные), тройки атомов (трехчастичные) и четверки атомов (четырехчастичные). Сложность алгоритма является линейной, что позволяет значительно понизить вычислительную сложность расчетов.
При оценке ViSNet использовались стандартизированные критерии для прогнозирования молекулярных свойств. На всех существующих датасетах (MD17, MD22, QM9 и Molecule3D) ViSNet превосходил существующие алгоритмы по точности определения геометрии молекул. Также ViSNet использовалась в первом глобальном конкурсе по разработке лекарств на основе искусственнго интеллекта, международном конкурсе по прогнозированию ингибиторов против основной протеазы SARS-CoV-2, учитывая информацию о последовательности малых молекул. Во всем мире в соревновании приняли участие 1105 участников из 878 команд. ViSNet победила в соревновании.
ViSNET добавлена в PyTorch в качестве базовой модели для молекулярного моделирования и прогнозирования свойств молекул. Также модель опубликована на GitHub.